The Novel route of investigation for the application of Quantum chemistry to clarify the new synthetic route for Dasatinib from (E)-Ethyl-3-ethoxy acrylate by using various reagents. The Overall Reaction carried out in Eight Steps. Which are less than earlier reported synthetic schemes. The Energy of every reactant, Intermediate and products were calculated by using DFT (Density Functional Theory). The energies diagram obtained shown the new proposed scheme could follow the easy path to obtain the product, moreover, the energy barrier required to overcome the transition state is low indicating, very less activation energy is required for every reactant to take part in chemical reaction. The energy diagram that was obtained shows that the new plan that was suggested could follow an easy path to obtaining Product.
| Published in | International Journal of Computational and Theoretical Chemistry (Volume 13, Issue 1) |
| DOI | 10.11648/j.ijctc.20251301.11 |
| Page(s) | 1-12 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Dasatinib, Quantum Chemistry, Transition State, Density Functional Theory (DFT), Cyclization, Halogenation, Quantum Chemistry (QM), Regioselective Demethylation
EDC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide |
HCl | Hydrochloric acid |
HOBt | Hydroxy benzotriazole |
Pd(dppf)Cl2 | [1,1'-Bis(diphenylphosphino) ferrocene] dichloropalladium(II) |
NaOtBu | Sodium tert-butoxide |
NaCl | Sodium chloride |
Serial No | Compound No | Energy (Kcal/mol) |
|---|---|---|
1 | Compound-I | -73.2 |
2 | Compound-II | -68.4 |
3 | TS-I | 104.2 |
4 | TS-II | 109.3 |
5 | INT-I | -60.1 |
6 | TS-III | 101.3 |
7 | TS-IV | 105.4 |
8 | TS-V | 102.5 |
9 | INT-II | -62.8 |
10 | TS-VI | 98.3 |
11 | INT-III | -46.3 |
12 | TS-VII | 99.3 |
13 | TS-VIII | 102.7 |
14 | Dasatinib | -78.6 |
Compounds | HOMO | LUMO | Energy(gap)LUMO -HOMO |
|---|---|---|---|
compound 1 | -0.23554 | -0.05918 | 0.17636 |
compound 2 | -0.19768 | 0.00252 | 0.2002 |
TS I | -0.31743 | -0.16193 | 0.1555 |
TS II | -0.23902 | -0.07729 | 0.16173 |
INT 1 | -0.22964 | -0.05255 | 0.17709 |
TS III | -0.30946 | -0.18901 | 0.12045 |
Int 2 | -0.2153 | -0.03904 | 0.17626 |
TS IV | -0.32895 | -0.2031 | 0.12585 |
TS V | -0.23375 | -0.07 | 0.16375 |
INT 3 | -0.23177 | -0.06553 | 0.16624 |
TS VI | -0.32861 | -0.191 | 0.13761 |
TS VII | -0.22388 | -0.0626 | 0.16128 |
TS VIII | -0.22386 | -0.06257 | 0.16129 |
Dasatinib | -0.20967 | -0.05586 | 0.15381 |
HOMO | Highest Occupied Molecular Orbital |
LUMO | Lowest Unoccupied Molecular Orbital |
| [1] | Thiazoles and Bisthiazoles [Internet]. [cited 2024 Sep 29]. Available from: |
| [2] |
Zoltewicz JA, Deady LW. Quaternization of Heteroaromatic Compounds: Quantitative Aspects. In: Katritzky AR, Boulton AJ, editors. Advances in Heterocyclic Chemistry [Internet]. Academic Press; 1978 [cited 2024 Sep 29]. p. 71–121. Available from:
https://www.sciencedirect.com/science/article/pii/S0065272508601038 |
| [3] | Thiazole Ring—A Biologically Active Scaffold [Internet]. [cited 2024 Sep 29]. Available from: |
| [4] | Thiazole: A Versatile Standalone Moiety Contributing to the Development of Various Drugs and Biologically Active Agents [Internet]. [cited 2024 Sep 29]. Available from: |
| [5] | Parikh A, Parikh H, Parikh K, editors. HantzschThiazole, Pyridine and 1,2,4-Triazine Synthesis. In: Name Reactions in Organic Synthesis [Internet]. Foundation Books; 2006 [cited 2024 Sep 29]. p. 544–6. Available from: |
| [6] |
318. Studies in the azole series. Part I. A novel route to 5-aminothiazoles - Journal of the Chemical Society (Resumed) (RSC Publishing) [Internet]. [cited 2024 Sep 29]. Available from:
https://pubs.rsc.org/en/content/articlelanding/1947/jr/jr9470001594 |
| [7] |
Kumdale, P. G., &Shitole, N. V. Glutamic Acid as an Efficient Catalyst Used for the Synthesis of 5-Arylidene-2, 4-Thiazolidinediones- Asian journal of Organic & medicinal chemistry, [7(2) cited 2022] Available from
https://scholar.google.com/scholar?oi=bibs&cluster=1771390021643789254&btnI=1&hl=en |
| [8] | (PDF) Microwave Assisted Synthesis of Five Membered Azaheterocyclic Systems [Internet]. [cited 2024 Sep 29]. Available from: |
| [9] | Kriek M, Martins F, Leonardi R, Fairhurst SA, Lowe DJ, Roach PL. Thiazole Synthase from Escherichia coli. Journal of Biological Chemistry. 2007 Jun; 282(24): 17413–23. |
| [10] | Fei Y, Chen Z, Zhang J, Yu M, Kong J, Wu Z, et al. Thiazolium-based ionic liquids: Synthesis, characterization and physicochemical properties. Journal of Molecular Liquids. 2021 Nov 15; 342: 117553. |
| [11] | Olivieri A, Manzione L. Dasatinib: a new step in molecular target therapy. Annals of Oncology. 2007 Jun; 18: vi42–6. |
| [12] | Keating GM. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs. 2017 Jan; 77(1): 85–96. |
| [13] | Chen BC, Zhao R, Wang B, Droghini R, Lajeunesse J, Sirard P, et al. A new and efficient preparation of 2-aminothiazole-5-carbamides: applications to the synthesis of the anti-cancer drug dasatinib. Stevens CV, editor. Arkivoc. 2009 Nov 25; 2010(6): 32–8. |
| [14] | Discovery of N-(2-Chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a Dual Src/Abl Kinase Inhibitor with Potent Antitumor Activity in Preclinical Assays | Journal of Medicinal Chemistry [Internet]. [cited 2024 Sep 29]. Available from: |
| [15] | Griffith JS, Orgel LE. Ligand-field theory. Q Rev Chem Soc. 1957 Jan 1; 11(4): 381–93. |
| [16] |
Homology Modeling - an overview | ScienceDirect Topics [Internet]. [cited 2024 Sep 25]. Available from:
https://www.sciencedirect.com/topics/biochemistry-genetics-and-molecular-biology/homology-modeling |
| [17] | Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals | Organic Process Research & Development [Internet]. [cited 2024 Sep 30]. Available from: |
| [18] | Kinetics and Mechanism of N-Boc Cleavage: Evidence of a Second-Order Dependence upon Acid Concentration | The Journal of Organic Chemistry [Internet]. [cited 2024 Sep 30]. Available from: |
| [19] |
Nucleophilic Aromatic Substitution (SNAr) as an Approach to Challenging Carbohydrate–Aryl Ethers - ScienceDirect [Internet]. [cited 2024 Sep 30]. Available from:
https://www.sciencedirect.com/org/science/article/pii/S1523706021115883 |
| [20] |
Dialkylbiaryl phosphines in Pd-catalyzed amination: a user’s guide - Chemical Science (RSC Publishing) [Internet]. [cited 2024 Sep 30]. Available from:
https://pubs.rsc.org/en/content/articlelanding/2011/sc/c0sc00331j |
APA Style
Kumdale, P., Chavan, A., Reddy, S. (2025). Computational Elucidation of Novel Synthetic Scheme for Dasatinib. International Journal of Computational and Theoretical Chemistry, 13(1), 1-12. https://doi.org/10.11648/j.ijctc.20251301.11
ACS Style
Kumdale, P.; Chavan, A.; Reddy, S. Computational Elucidation of Novel Synthetic Scheme for Dasatinib. Int. J. Comput. Theor. Chem. 2025, 13(1), 1-12. doi: 10.11648/j.ijctc.20251301.11
AMA Style
Kumdale P, Chavan A, Reddy S. Computational Elucidation of Novel Synthetic Scheme for Dasatinib. Int J Comput Theor Chem. 2025;13(1):1-12. doi: 10.11648/j.ijctc.20251301.11
@article{10.11648/j.ijctc.20251301.11,
author = {Prashant Kumdale and Arun Chavan and Sanjeev Reddy},
title = {Computational Elucidation of Novel Synthetic Scheme for Dasatinib
},
journal = {International Journal of Computational and Theoretical Chemistry},
volume = {13},
number = {1},
pages = {1-12},
doi = {10.11648/j.ijctc.20251301.11},
url = {https://doi.org/10.11648/j.ijctc.20251301.11},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijctc.20251301.11},
abstract = {The Novel route of investigation for the application of Quantum chemistry to clarify the new synthetic route for Dasatinib from (E)-Ethyl-3-ethoxy acrylate by using various reagents. The Overall Reaction carried out in Eight Steps. Which are less than earlier reported synthetic schemes. The Energy of every reactant, Intermediate and products were calculated by using DFT (Density Functional Theory). The energies diagram obtained shown the new proposed scheme could follow the easy path to obtain the product, moreover, the energy barrier required to overcome the transition state is low indicating, very less activation energy is required for every reactant to take part in chemical reaction. The energy diagram that was obtained shows that the new plan that was suggested could follow an easy path to obtaining Product.
},
year = {2025}
}
TY - JOUR T1 - Computational Elucidation of Novel Synthetic Scheme for Dasatinib AU - Prashant Kumdale AU - Arun Chavan AU - Sanjeev Reddy Y1 - 2025/02/26 PY - 2025 N1 - https://doi.org/10.11648/j.ijctc.20251301.11 DO - 10.11648/j.ijctc.20251301.11 T2 - International Journal of Computational and Theoretical Chemistry JF - International Journal of Computational and Theoretical Chemistry JO - International Journal of Computational and Theoretical Chemistry SP - 1 EP - 12 PB - Science Publishing Group SN - 2376-7308 UR - https://doi.org/10.11648/j.ijctc.20251301.11 AB - The Novel route of investigation for the application of Quantum chemistry to clarify the new synthetic route for Dasatinib from (E)-Ethyl-3-ethoxy acrylate by using various reagents. The Overall Reaction carried out in Eight Steps. Which are less than earlier reported synthetic schemes. The Energy of every reactant, Intermediate and products were calculated by using DFT (Density Functional Theory). The energies diagram obtained shown the new proposed scheme could follow the easy path to obtain the product, moreover, the energy barrier required to overcome the transition state is low indicating, very less activation energy is required for every reactant to take part in chemical reaction. The energy diagram that was obtained shows that the new plan that was suggested could follow an easy path to obtaining Product. VL - 13 IS - 1 ER -
Department of Chemistry, Shivneri College Shirur Anantpal Dist, Latur, India
Department of Chemistry, School of Chemical Sciences, Swami Ramanand Teerth Marathwada University, Nanded, India
Department of Chemistry, Gramin ACS Mahavidyalaya, Vasant Nagar, Mukhed, India
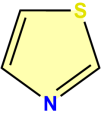
Figure 1. Structure of 1, 3-Thiazole.
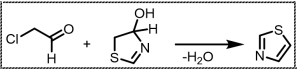
Scheme 1. Hantzch method for thiazole synthesis.
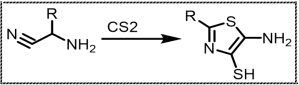
Scheme 2. Cook-Heilbron method for thiazole synthesis.
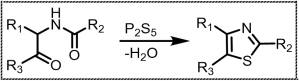
Scheme 3. Gabriel method for thiazole synthesis.
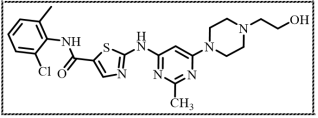
Figure 2. Structure of N- (2- chloro -6 -methylphenyl)- 2-((6- (4- (2-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-yl)amino)thiazole-5-carboxamide.
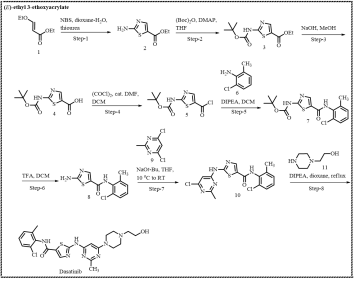
Scheme 4. Novel synthetic route reported by Bristol-Myers Squibb, USA.
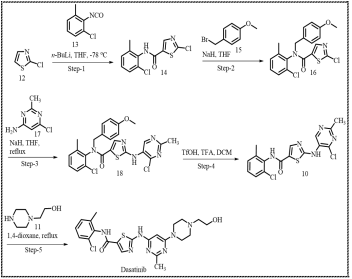
Scheme 5. Robust and scalable synthetic route reported by Louis J. Lombardo et al.
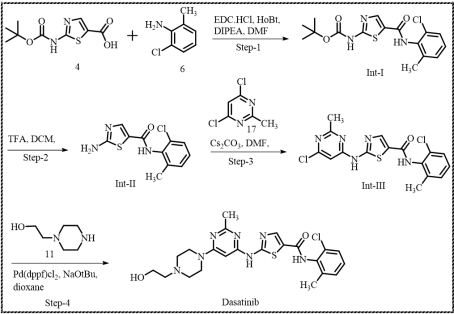
Scheme 6. Proposed robust and scalable synthetic route for the Dasatinib.
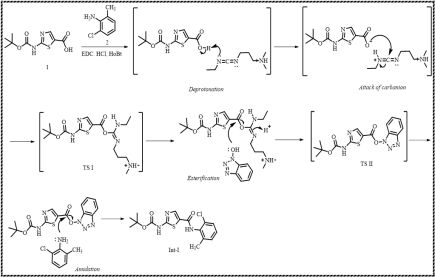
Scheme 7. Formation of Intermediate-I (Int-I).
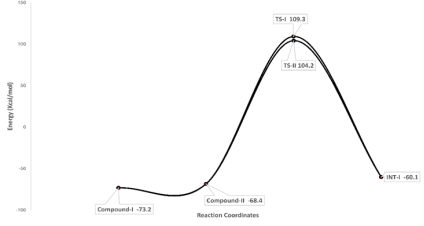
Figure 3. Energy profile diagram for the formation of Int-I.
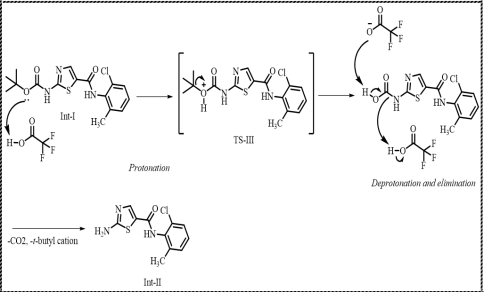
Scheme 8. Formation of Intermediate-II.
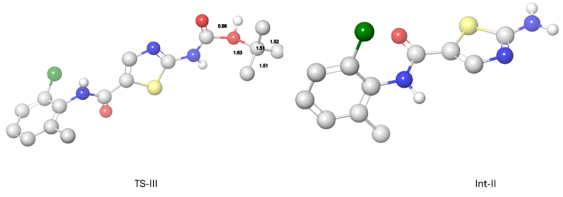
Figure 5. Optimal atomic distance between the reactant atoms in TS-III and Int-II.
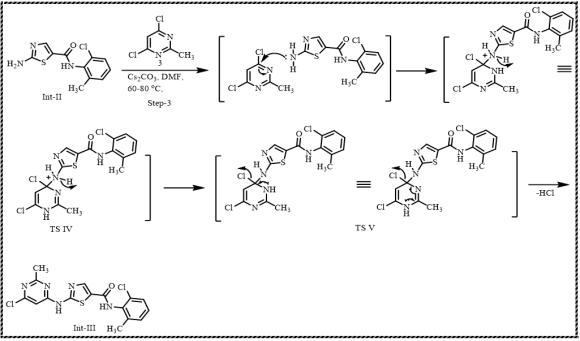
Scheme 9. Formation of Intermediate-III.
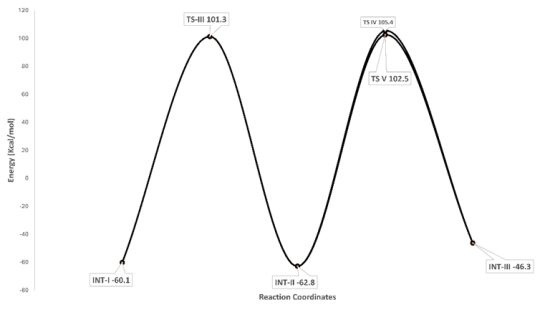
Figure 6. Potential energy profile diagram for the formation of Int-II and Int-III.
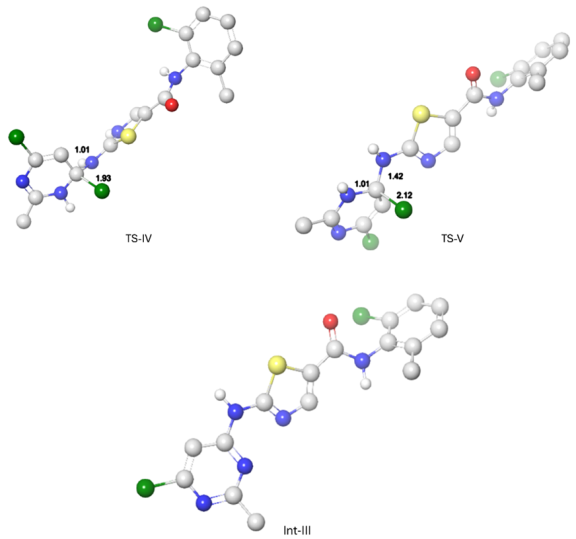
Figure 7. Optimal atomic distance between the reactant atoms in TS-IV, TS-V and Int-III.
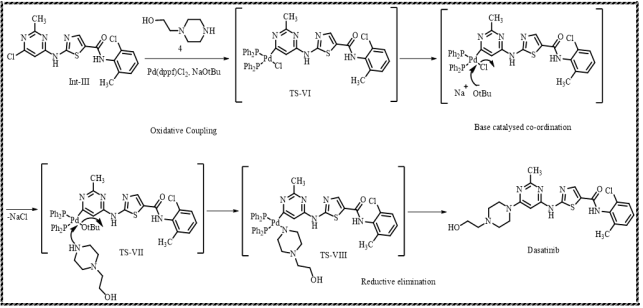
Scheme 10. Formation of Dasatinib from Int-III.
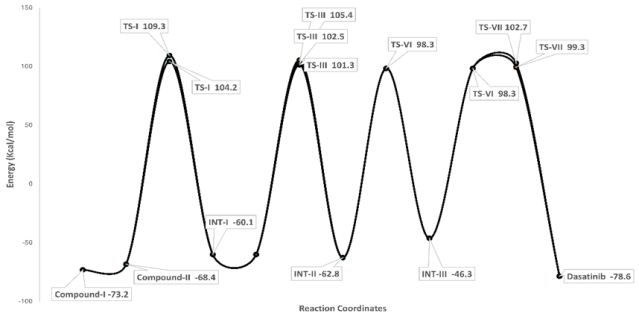
Figure 8. Over all energy profile diagram for the formation of Dasatinib.
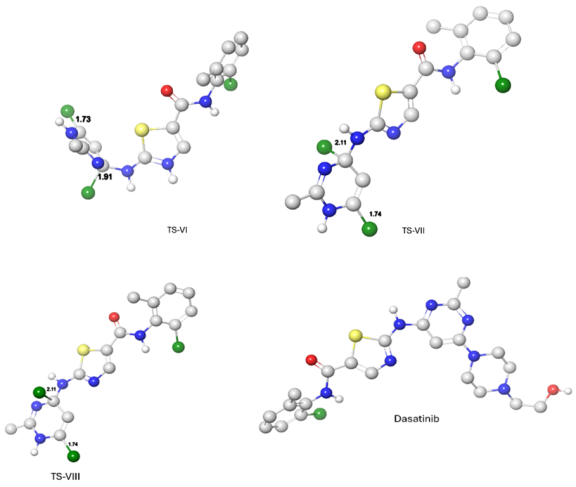
Figure 9. Optimal atomic distance between the reactant atoms in TS-V, TS-VII, TS-VIII and Dasatinib.
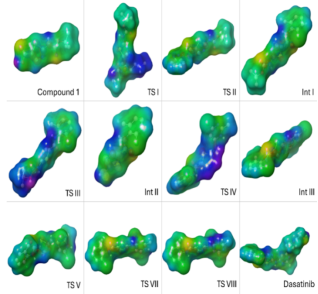
Figure 10. HOMO- LUMO and potential energy surface for compounds, intermediates and Transition states.
Information

