This groundbreaking research rigorously investigated the CO2 absorption potential of two potassium-based ionic liquids (ILs), namely potassium benzene disulfonamide [C6H4KNS2O4] and potassium phthalimide [C8H4KNO2]. Driven by the urgent need for effective carbon capture technologies to combat climate change stemming from fossil fuel combustion, this study employed sophisticated Density Functional Theory (DFT) calculations using the M062X/6-31+G(d,p) method. The computational approach encompassed comprehensive geometry optimization, in-depth molecular interaction analyses, precise binding energy assessments, insightful Natural Bond Orbital (NBO) analysis, and a thorough evaluation of solvent effects. The findings unequivocally demonstrate that both ILs exhibit tangible interactions with CO2, with binding energies ranging from -3.108 to -0.232 kcal/mol for C6H4KNS2O4 and -3.475 to -0.219 kcal/mol for C8H4KNO2. These energies strongly suggest the viability of these ILs for CO2 capture applications, potentially requiring minimal energy for regeneration. Crucially, the research established that potassium benzene disulfonamide [C6H4KNS2O4] displays superior CO2 capture efficacy compared to potassium phthalimide [C8H4KNO2]. This conclusion is robustly supported by compelling thermochemical and molecular interaction data. NBO analysis further elucidated that CO2 interaction induces alterations in the IL geometry and facilitates charge transfer between the interacting species. Moreover, studies on cation-anion interactions revealed a stronger association between C6H4KNS2O4 and the potassium cation (K+). Investigation of isolated anion interactions with CO2 echoed the preference for [C6H4NS2O4]. While solvent effects influenced thermochemical properties, they did not fundamentally alter the geometry of the anion-CO2 complexes. In conclusion, the computational evidence unequivocally indicates the formation of stable complexes between the investigated IL pairs and CO2 molecules. Most significantly, this study firmly establishes that C6H4KNS2O4 is a more promising candidate for efficient CO2 absorption, offering a pathway towards the development of advanced and effective CO2 capture technologies.
| Published in | International Journal of Computational and Theoretical Chemistry (Volume 13, Issue 1) |
| DOI | 10.11648/j.ijctc.20251301.13 |
| Page(s) | 25-42 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Absorption, Binding Energy, Carbon Dioxide, DFT, Ionic LIquid, NBO Analysis
Ionic liquids | ||
|---|---|---|
Thermochemical data | C6H4KNS2O4 | C8H4KNO2 |
∆B.E, kcal/ mol | -127.178 | -115.364 |
Dipole moment, Debye | 7.2959 | 7.8982 |
(a) | [C6H4NS2O4]-CO2 complexes | ∆B.E | ∆H | ∆S | Dpm |
[C6H4NS2O4]-CO2 I (a) | -37.025 | -37.61 | -28.863 | 6.42 | |
[C6H4NS2O4]-CO2 II (b) | -39.274 | -39.86 | -30.959 | 8.86 | |
[C6H4NS2O4]-CO2 III (c) | -39.2564 | -39.84 | -32.124 | 8.28 | |
[C6H4NS2O4]-CO2 IV(d) | -39.26 | -39.85 | -31.282 | 8.67 | |
(b) | [C8H4NO2]-CO2 complexes | ∆B.E | ∆H | ∆S | Dpm |
[C8H4NO2]-CO2I (e) | -24.186 | -24.77 | -32.785 | 13.26 | |
[C8H4NO2]-CO2 II (f) | 23.336 | 22.744 | -36.702 | 9.442 | |
[C8H4NO2]-CO2 III(g) | -21.177 | -21.77 | -23.228 | 10.9 |
Complexes | Charge distribution | Bending angle | |
|---|---|---|---|
(a’) | [C6H4NS2O4]-CO2 I (a) | -1.465 | 174.777 |
[C6H4NS2O4]-CO2 II (b) | -1.481 | 174.684 | |
[C6H4NS2O4]-CO2 III(c) | -1.481 | 174.70091 | |
[C6H4NS2O4]-CO2 IV (d) | -1.639 | 175.818 | |
(b’) | [C8H4NO2]-CO2 I (e) | -0.68 | 138.37007 |
[C8H4NO2]-CO2 II (f) | -0.579 | 120.61532 | |
[C8H4NO2]-CO2 III (g) | -0.689 | 177.29593 |
Thermodynamic values | C6H4KNS2O4 -CO2 Conformer | C8H4KNO2 -CO2 Conformer | ||||
|---|---|---|---|---|---|---|
(a) | (b) | (c ) | (d) | (e) | (f) | |
C−O, Å | 1.1544 | 1.1561 | 1.165 | 1.155 | 1.156 | 1.17 |
< O-C-O, 0 | 176.53 | 179.92 | 177.13 | 175.45 | 179.93 | 174.5 |
Relative energy, kcal mol-1 | 1.0000 | 0.000 | 1.000 | 1.000 | 1.000 | 0.000 |
∆B.E, kcal mol-1 | -25.989 | -21.585 | -23.58 | -26.15 | -21.42 | -25.85 |
∆H, kcal mol-1 | -26.5813 | -22.177 | -24.172 | -26.74 | -22.01 | -26.44 |
∆S, cal mol-1k-1 | -27.406 | -21.497 | -28.88 | -28.00 | -20.22 | -25.18 |
Dpm, Debye | 6.44 | 8.07 | 6.93 | 7.15 | 9.05 | 7.04 |
Species | Donor(i) | Accepter(j) | E2(kcal/mol) | Ej-Ei/a.u |
|---|---|---|---|---|
C8H4KNO2 | LP1(N13) | BD*1(C2-C8) | 11.45 | 1.24 |
LP2(O14) | BD*1(C2-C8) | 25.88 | 1.12 | |
LP2(O14) | BD*1(C8-N13) | 24.20 | 1.23 | |
LP2(O14) | BD*1(C8-N13) | 24.20 | 1.23 | |
LP2(O 14) | BD*1(C8-N13) | 24.20 | 1.23 | |
LP2(O15) | BD*1(C7-N13) | 32.00 | 1.16 | |
CO2/C8H4KNO2 | LP2(O14) | BD*1(C2-C8) | 26.15 | 1.12 |
LP2(O14) | BD*1(C8-N13) | 24.51 | 1.23 | |
LP2(O15) | BD*1(C1-C7) | 28.81 | 1.09 | |
LP2(O15) | BD*1(C7-N13) | 31.88 | 1.16 | |
LP3(O15) | BD*2(C7-N13) | 266.99 | 0.42 | |
C6H4KNS2O4 | LP2(N13) | BD*1(S11-O15) | 18.54 | 0.62 |
LP2(O14) | BD*1(S11-O15) | 30.66 | 0.66 | |
LP3(O14) | BD*1(C2-S11) | 22.13 | 0.54 | |
LP2(O15) | BD*1(S11-N13) | 22.69 | 0.63 | |
LP3(O15) | BD*1(C2-S11) | 20.46 | 0.56 | |
LP2(O16) | BD*1(S12-N13) | 22.70 | 0.63 | |
LP3(O16) | BD*1(C1-S12) | 20.47 | 0.56 | |
LP2(O17) | BD*1(S12-O16) | 30.66 | 0.66 | |
LP3(O17) | BD*1(C1-S12) | 22.12 | 0.54 | |
LP3(O17) | BD*1(S12-N13) | 21.52 | 0.60 | |
CO2/C6H4KNS2O4 | LP2(O14) | BD*1(S11-O15) | 33.06 | 0.96 |
LP3(O14) | BD*1(C2-S11) | 24.37 | 0.80 | |
LP3(O14) | BD*1(S11-N13) | 23.38 | 0.87 | |
LP2(O15) | BD*1(S11-N13) | 25.39 | 0.90 | |
LP3(O15) | BD*1(C2-S11) | 22.66 | 0.82 | |
LP2(O16) | BD*1(S12-N13) | 25.72 | 0.90 | |
LP3(O16) | BD*1(C1-S12) | 22.22 | 0.82 | |
LP2(O17) | BD*1(S12-O16) | 32.40 | 0.96 | |
LP3(O17) | BD*1(C1-S12) | 26.73 | 0.80 | |
LP3(O17) | BD*1(S12-N13) | 20.09 | 0.87 |
(a) [C8H4NO2] - CO2 complexes | |||||||||
Chloroform | DMSO | Water | |||||||
∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | |
I | -7.26 | -6.67 | -35.1 | -8.08 | -7.19 | -35.55 | -8.13 | -7.54 | -35.54 |
II | 13.74 | 14.3 | -36.21 | 14.47 | 15.06 | -36.43 | 14.48 | 15.07 | -36.35 |
III | -1.73 | -1.139 | -24.04 | -1.51 | -0.92 | -23.09 | -1.5 | -0.913 | -22.63 |
(b) [C6H4NS2O4] - CO2 complexes | |||||||||
Chloroform | DMSO | Water | |||||||
∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | |
I | -3.01 | -2.42 | -29.53 | -2.7 | -2.11 | -26.51 | -2.69 | -2.10 | -26.25 |
II | -3.8 | -3.21 | -29.33 | -3.04 | -2.45 | -27.69 | -3.009 | -2.41 | -27.74 |
III | -3.81 | -3.22 | -27.099 | -3.003 | -2.41 | -27.99 | -2.97 | -2.377 | -28.13 |
IV | -3.22 | -2.634 | -29.84 | -2.94 | -2.35 | -29.12 | -2.94 | -2.35 | -29.17 |
Chloroform | DMSO | Water | |||||||
|---|---|---|---|---|---|---|---|---|---|
ILs | ∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S |
C6H4KNS2O4 | -28.11 | -20.7 | -26.5 | -8.806 | -8.21 | -25.46 | -7.91 | -7.32 | -25.48 |
C8H4KNO2 | -30.04 | -29.45 | -24.0 | -9.69 | -9.10 | -23.11 | -8.78 | -8.19 | -23.05 |
Chloroform | DMSO | Water | |||||||
|---|---|---|---|---|---|---|---|---|---|
∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | ∆H | ∆B.E | ∆S | |
(a) | -3.69 | -3.108 | -24.98 | -3.05 | -2.465 | -26.63 | -3.02 | -2.435 | -26.66 |
(b) | -0.82 | -0.232 | -21.40 | -0.049 | 0.542 | -18.62 | 0.025 | 0.618 | -20.56 |
(c) | -3.51 | -2.922 | -26.40 | -2.828 | -2.235 | -25.97 | -2.81 | -2.221 | -26.89 |
(d) | -4.06 | -3.475 | -27.39 | -3.23 | -2.644 | -27.19 | -3.19 | -2.605 | -28.79 |
(e) | -0.811 | -0.219 | -19.12 | 0.075 | 0.668 | -19.65 | 0.11 | 0.703 | -19.62 |
(f) | -2.809 | -2.218 | -22.83 | -1.65 | -1.063 | -22.84 | -1.6 | -1.014 | -23.17 |
DFT | Density Functional Theory |
DMSO | Dimethyl Sulfoxide |
E2 | Second-Order Perturbation Stabilization Energies |
ESP | Electrostatic Potential |
HOMO | Highest Occupied Molecular Orbital |
ILs | Ionic Liquids |
LUMO | Lowest Unoccupied Molecular Orbital |
MEA | Monoethanol Amine |
MESP | Molecular Electrostatic Potential |
NBO | Natural Bond Orbital |
PCM | Polarizable Continuum Model |
SCRF | Self-Consistent Reaction Field |
| [1] | Y. Zhang, X. Lu and X. Ji, Carbon dioxide capture, Deep Eutectic Solvents Synth. Prop. Appl. (2019), pp. 297-319. |
| [2] | S. Harrison, J. Franklin, R. R. Hernandez, M. Ikegami, H. D. Safford and J. H. Thorne, Climate change and California’s terrestrial biodiversity, Proc. Natl. Acad. Sci. 121 (2024), pp. e2310074121. |
| [3] | T. M. Gür, Carbon dioxide emissions, capture, storage and utilization: Review of materials, processes and technologies, Prog. Energy Combust. Sci. 89 (2022), pp. 100965. |
| [4] | M. C. Stern, F. Simeon, H. Herzog and T. A. Hatton, Post-combustion carbon dioxide capture using electrochemically mediated amine regeneration, Energy Environ. Sci. 6 (2013), pp. 2505-2517. |
| [5] | F. Wang, S. Deng, H. Zhang, J. Wang, J. Zhao, H. Miao et al., A comprehensive review on high-temperature fuel cells with carbon capture, Appl. Energy 275 (2020), pp. 115342. |
| [6] | M. Freemantle, Ionic liquids may boost clean technology development, Chem Eng News 76 (1998), pp. 32-37. |
| [7] | X. Fan, S. Liu, Z. Jia, J. J. Koh, J. C. C. Yeo, C.-G. Wang et al., Ionogels: recent advances in design, material properties and emerging biomedical applications, Chem. Soc. Rev. 52 (2023), pp. 2497-2527. |
| [8] | J. Cao, D. Zhang, X. Zhang, Z. Zeng, J. Qin and Y. Huang, Strategies of regulating Zn 2+ solvation structures for dendrite-free and side reaction-suppressed zinc-ion batteries, Energy Environ. Sci. 15 (2022), pp. 499-528. |
| [9] | L. A. Blanchard, D. Hancu, E. J. Beckman and J. F. Brennecke, Green processing using ionic liquids and CO2, Nature 399 (1999), pp. 28-29. |
| [10] | B. Xue, Y. Yu, J. Chen, X. Luo and M. Wang, A comparative study of MEA and DEA for post-combustion CO 2 capture with different process configurations, Int. J. Coal Sci. Technol. 4 (2017), pp. 15-24. |
| [11] | F. Vega, S. Camino, J. Camino, J. Garrido and B. Navarrete, Partial oxy-combustion technology for energy efficient CO2 capture process, Appl. Energy 253 (2019), pp. 113519. |
| [12] | F. U. Shah, R. An and N. Muhammad, Properties and applications of ionic liquids in energy and environmental science, Front. Chem. 8 (2020), pp. 627213. |
| [13] | D. Bálint and L. Jäntschi, Comparison of molecular geometry optimization methods based on molecular descriptors, Mathematics 9 (2021), pp. 2855. |
| [14] | J. J. Stewart, Application of the PM6 method to modeling the solid state, J. Mol. Model. 14 (2008), pp. 499-535. |
| [15] | N. S. Babu, Applications of Current Density Functional Theory (DFT) Methods in Polymer Solar Cells, in Density Functional Theory-Recent Advances, New Perspectives and Applications, IntechOpen, 2021. |
| [16] | V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, 6-31G* basis set for third-row atoms, J. Comput. Chem. 22 (2001), pp. 976-984. |
| [17] | Y. Sert, L. Singer, M. Findlater, H. Doğan and Ç. Çırak, Vibrational frequency analysis, FT-IR, DFT and M06-2X studies on tert-Butyl N-(thiophen-2yl) carbamate, Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 128 (2014), pp. 46-53. |
| [18] | M. Mercy, N. H. de Leeuw and R. G. Bell, Mechanisms of CO 2 capture in ionic liquids: a computational perspective, Faraday Discuss. 192 (2016), pp. 479-492. |
| [19] | B. Lai and C. Oostenbrink, Binding free energy, energy and entropy calculations using simple model systems, Theor. Chem. Acc. 131 (2012), pp. 1-13. |
| [20] | R. Gangadharan and S. Sampath Krishnan, Natural Bond Orbital (NBO) population analysis of 1-azanapthalene-8-ol, Acta Phys. Pol. A 125 (2014), pp. 18-22. |
| [21] | B. Cao, J. Du, S. Liu, X. Zhu, X. Sun, H. Sun et al., Carbon dioxide capture by amino-functionalized ionic liquids: DFT based theoretical analysis substantiated by FT-IR investigation, RSC Adv. 6 (2016), pp. 10462-10470. |
| [22] | K. Dhar and S. Fahim, Investigation of the Absorption of CO2 in Ionic Liquid, Bangladesh J. Sci. Res. 29 (2016), pp. 41-46. |
| [23] | D. Josa, J. Rodríguez-Otero, E. M. Cabaleiro-Lago and M. Rellán-Piñeiro, Analysis of the performance of DFT-D, M05-2X and M06-2X functionals for studying π⋯ π interactions, Chem. Phys. Lett. 557 (2013), pp. 170-175. |
| [24] | Y. Wang, P. Verma, X. Jin, D. G. Truhlar and X. He, Revised M06 density functional for main-group and transition-metal chemistry, Proc. Natl. Acad. Sci. 115 (2018), pp. 10257-10262. |
| [25] | V. Lachet, T. de Bruin, P. Ungerer, C. Coquelet, A. Valtz, V. Hasanov et al., Thermodynamic behavior of the CO2+ SO2 mixture: Experimental and Monte Carlo simulation studies, Energy Procedia 1 (2009), pp. 1641-1647. |
| [26] | M. B. Shiflett and Ajj. Yokozeki, Phase behavior of carbon dioxide in ionic liquids: [emim][acetate], [emim][trifluoroacetate], and [emim][acetate]+[emim][trifluoroacetate] mixtures, J. Chem. Eng. Data 54 (2009), pp. 108-114. |
| [27] | V. E. Romanovsky, D. Drozdov, N. G. Oberman, G. Malkova, A. L. Kholodov, S. Marchenko et al., Thermal state of permafrost in Russia, Permafr. Periglac. Process. 21 (2010), pp. 136-155. |
| [28] | B. Shimekit and H. Mukhtar, Natural gas purification technologies-major advances for CO2 separation and future directions, Adv. Nat. Gas Technol. 2012 (2012), pp. 235-270. |
| [29] | J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Muller et al., Combining machine learning and computational chemistry for predictive insights into chemical systems, Chem. Rev. 121 (2021), pp. 9816-9872. |
| [30] | H. Sun, B. Cao, Q. Tian, S. Liu, D. Du, Z. Xue et al., A DFT study on the absorption mechanism of vinyl chloride by ionic liquids, J. Mol. Liq. 215 (2016), pp. 496-502. |
| [31] | Mukhtar, S. Saqib, N. B. Mellon, M. Babar, S. Rafiq, S. Ullah et al., CO2 capturing, thermo-kinetic principles, synthesis and amine functionalization of covalent organic polymers for CO2 separation from natural gas: A review, J. Nat. Gas Sci. Eng. 77 (2020), pp. 103203. |
| [32] | M. Vafaeezadeh, J. Aboudi and M. M. Hashemi, A novel phenolic ionic liquid for 1.5 molar CO 2 capture: combined experimental and DFT studies, RSC Adv. 5 (2015), pp. 58005-58009. |
| [33] | G. Wu, Y. Liu, G. Liu and X. Pang, The CO2 absorption in flue gas using mixed ionic liquids, Molecules 25 (2020), pp. 1034. |
| [34] | E. Tílvez, N. Díaz, M. I. Menéndez, D. Suárez and R. López, Quantum chemical calculations of stability constants: study of ligand effects on the relative stability of Pd (II)-peptide complexes, Theor. Chem. Acc. 128 (2011), pp. 465-475. |
APA Style
Tibebu, B., Geremu, A., Tsegaye, E. (2025). DFT Study on Potassium Benzene Disulfonamide and Potassium Phthalimide Ionic Liquid Based Carbon Dioxide Absorption. International Journal of Computational and Theoretical Chemistry, 13(1), 25-42. https://doi.org/10.11648/j.ijctc.20251301.13
ACS Style
Tibebu, B.; Geremu, A.; Tsegaye, E. DFT Study on Potassium Benzene Disulfonamide and Potassium Phthalimide Ionic Liquid Based Carbon Dioxide Absorption. Int. J. Comput. Theor. Chem. 2025, 13(1), 25-42. doi: 10.11648/j.ijctc.20251301.13
AMA Style
Tibebu B, Geremu A, Tsegaye E. DFT Study on Potassium Benzene Disulfonamide and Potassium Phthalimide Ionic Liquid Based Carbon Dioxide Absorption. Int J Comput Theor Chem. 2025;13(1):25-42. doi: 10.11648/j.ijctc.20251301.13
@article{10.11648/j.ijctc.20251301.13,
author = {Berihun Tibebu and Abdudin Geremu and Endale Tsegaye},
title = {DFT Study on Potassium Benzene Disulfonamide and Potassium Phthalimide Ionic Liquid Based Carbon Dioxide Absorption
},
journal = {International Journal of Computational and Theoretical Chemistry},
volume = {13},
number = {1},
pages = {25-42},
doi = {10.11648/j.ijctc.20251301.13},
url = {https://doi.org/10.11648/j.ijctc.20251301.13},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ijctc.20251301.13},
abstract = {This groundbreaking research rigorously investigated the CO2 absorption potential of two potassium-based ionic liquids (ILs), namely potassium benzene disulfonamide [C6H4KNS2O4] and potassium phthalimide [C8H4KNO2]. Driven by the urgent need for effective carbon capture technologies to combat climate change stemming from fossil fuel combustion, this study employed sophisticated Density Functional Theory (DFT) calculations using the M062X/6-31+G(d,p) method. The computational approach encompassed comprehensive geometry optimization, in-depth molecular interaction analyses, precise binding energy assessments, insightful Natural Bond Orbital (NBO) analysis, and a thorough evaluation of solvent effects. The findings unequivocally demonstrate that both ILs exhibit tangible interactions with CO2, with binding energies ranging from -3.108 to -0.232 kcal/mol for C6H4KNS2O4 and -3.475 to -0.219 kcal/mol for C8H4KNO2. These energies strongly suggest the viability of these ILs for CO2 capture applications, potentially requiring minimal energy for regeneration. Crucially, the research established that potassium benzene disulfonamide [C6H4KNS2O4] displays superior CO2 capture efficacy compared to potassium phthalimide [C8H4KNO2]. This conclusion is robustly supported by compelling thermochemical and molecular interaction data. NBO analysis further elucidated that CO2 interaction induces alterations in the IL geometry and facilitates charge transfer between the interacting species. Moreover, studies on cation-anion interactions revealed a stronger association between C6H4KNS2O4 and the potassium cation (K+). Investigation of isolated anion interactions with CO2 echoed the preference for [C6H4NS2O4]. While solvent effects influenced thermochemical properties, they did not fundamentally alter the geometry of the anion-CO2 complexes. In conclusion, the computational evidence unequivocally indicates the formation of stable complexes between the investigated IL pairs and CO2 molecules. Most significantly, this study firmly establishes that C6H4KNS2O4 is a more promising candidate for efficient CO2 absorption, offering a pathway towards the development of advanced and effective CO2 capture technologies.
},
year = {2025}
}
TY - JOUR T1 - DFT Study on Potassium Benzene Disulfonamide and Potassium Phthalimide Ionic Liquid Based Carbon Dioxide Absorption AU - Berihun Tibebu AU - Abdudin Geremu AU - Endale Tsegaye Y1 - 2025/04/10 PY - 2025 N1 - https://doi.org/10.11648/j.ijctc.20251301.13 DO - 10.11648/j.ijctc.20251301.13 T2 - International Journal of Computational and Theoretical Chemistry JF - International Journal of Computational and Theoretical Chemistry JO - International Journal of Computational and Theoretical Chemistry SP - 25 EP - 42 PB - Science Publishing Group SN - 2376-7308 UR - https://doi.org/10.11648/j.ijctc.20251301.13 AB - This groundbreaking research rigorously investigated the CO2 absorption potential of two potassium-based ionic liquids (ILs), namely potassium benzene disulfonamide [C6H4KNS2O4] and potassium phthalimide [C8H4KNO2]. Driven by the urgent need for effective carbon capture technologies to combat climate change stemming from fossil fuel combustion, this study employed sophisticated Density Functional Theory (DFT) calculations using the M062X/6-31+G(d,p) method. The computational approach encompassed comprehensive geometry optimization, in-depth molecular interaction analyses, precise binding energy assessments, insightful Natural Bond Orbital (NBO) analysis, and a thorough evaluation of solvent effects. The findings unequivocally demonstrate that both ILs exhibit tangible interactions with CO2, with binding energies ranging from -3.108 to -0.232 kcal/mol for C6H4KNS2O4 and -3.475 to -0.219 kcal/mol for C8H4KNO2. These energies strongly suggest the viability of these ILs for CO2 capture applications, potentially requiring minimal energy for regeneration. Crucially, the research established that potassium benzene disulfonamide [C6H4KNS2O4] displays superior CO2 capture efficacy compared to potassium phthalimide [C8H4KNO2]. This conclusion is robustly supported by compelling thermochemical and molecular interaction data. NBO analysis further elucidated that CO2 interaction induces alterations in the IL geometry and facilitates charge transfer between the interacting species. Moreover, studies on cation-anion interactions revealed a stronger association between C6H4KNS2O4 and the potassium cation (K+). Investigation of isolated anion interactions with CO2 echoed the preference for [C6H4NS2O4]. While solvent effects influenced thermochemical properties, they did not fundamentally alter the geometry of the anion-CO2 complexes. In conclusion, the computational evidence unequivocally indicates the formation of stable complexes between the investigated IL pairs and CO2 molecules. Most significantly, this study firmly establishes that C6H4KNS2O4 is a more promising candidate for efficient CO2 absorption, offering a pathway towards the development of advanced and effective CO2 capture technologies. VL - 13 IS - 1 ER -
Department of Chemistry, Jinka University, Jinka, Ethiopia
Department of Chemistry, Hawassa University, Hawassa, Ethiopia
Department of Chemistry, Hawassa University, Hawassa, Ethiopia
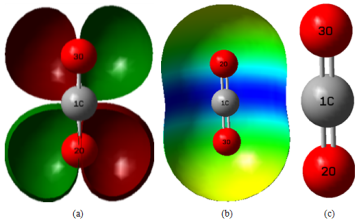
Figure 1. (a), HOMO-LUMO, (b) ESP map and (c), Optimized structures of CO2 calculated at M062X/6-31+G(d,p) level in gas phase.
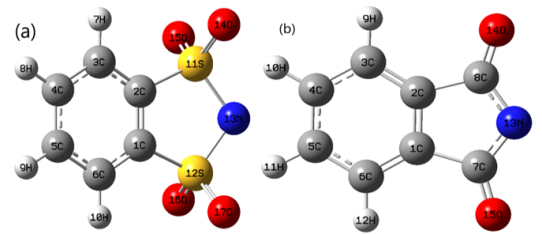
Figure 2. The geometric parameters for the optimized structures of (a) for [C6H4NS2O4] and (b) for [C8H4NO2] anions calculated at M062X/6-31+G(d,p) level in gas phase.
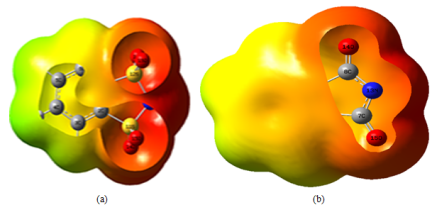
Figure 3. ESP map of (a) for [C6H4NS2O4] and (b) for [C8H4NO2] anions calculated at M062X/6-31+G(d,p) level in gas phase.
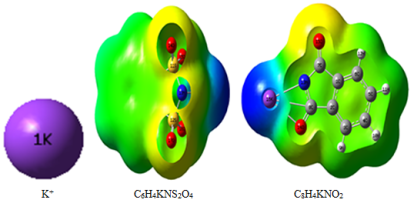
Figure 4. ESP surface of ILs and the optimized geometry of ion (K+) calculated at M062X/6-31+G(d,p) level in gas phase.
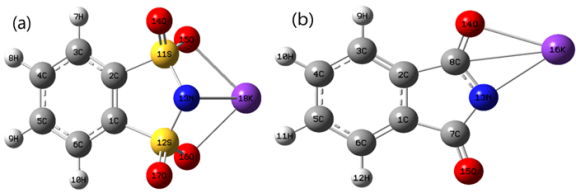
Figure 5. Ionic pairs comprising a potassium ion, [K+] with two anion calculated using M062X/6-31+G(d,p) level in gas phase.
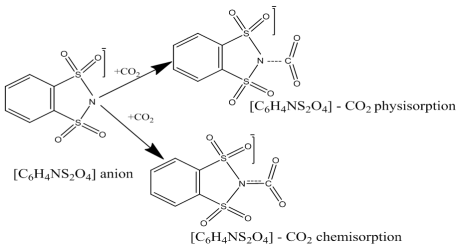
Scheme 1. Physisorption and Chemisorption mechanisms of [C6H4NS2O4] - CO2 from CO2 and N- interaction, structure of N-heterocyclic [C6H4NS2O4] - anion.
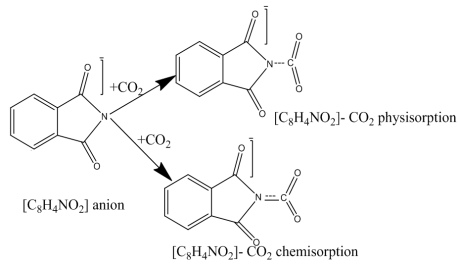
Scheme 2. The physisorption and chemisorption interaction mechanism between CO2 and [C8H4NO2] anion.
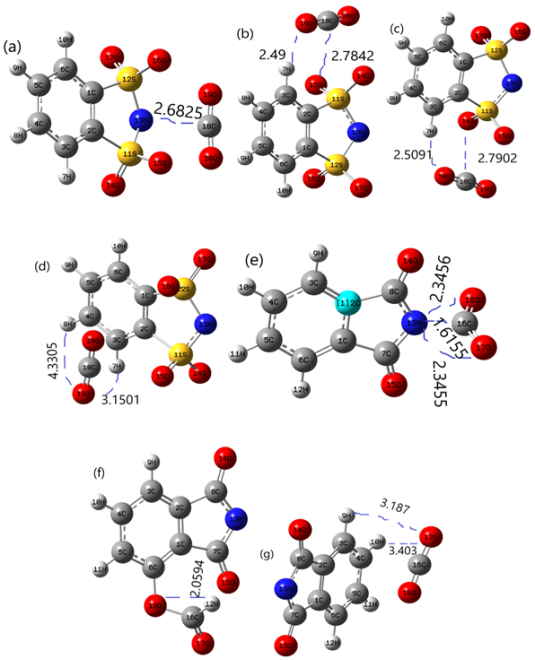
Figure 6. The optimized structures of [C8H4NO2]-CO2 and [C6H4NS2O4]-CO2 complexes in gas phase calculated at M062X/6-31+G (d, p) level.
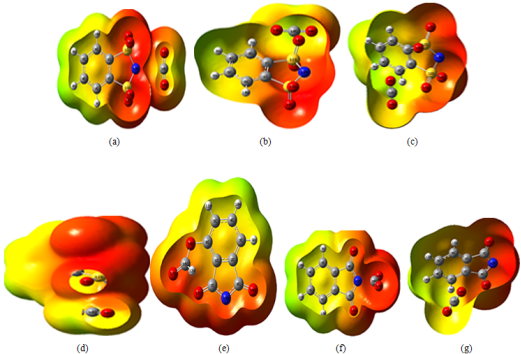
Figure 7. The ESP map of [C8H4NO2]-CO2 and [C6H4NS2O4]-CO2 complexes in the gas phase calculated at M062X/6-31+G (d, p) level.
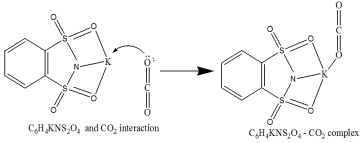
Scheme 3. The interaction mechanism between C6H4KNS2O4 and CO2.
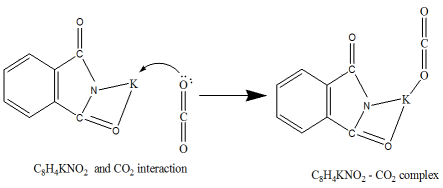
Scheme 4. The interaction mechanism between C8H4KNO2 with CO2.
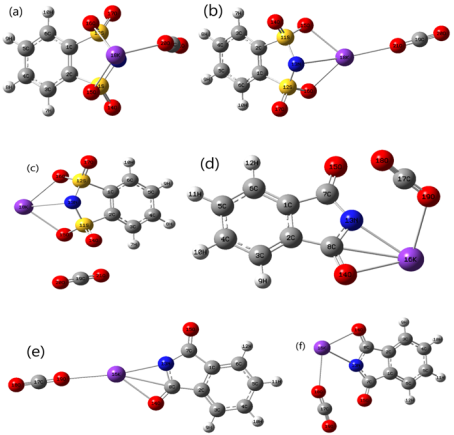
Figure 8. Optimized minimum energy structural IL-CO2 complexes are calculated at M062X/6-31+G(d,p) level in gas.
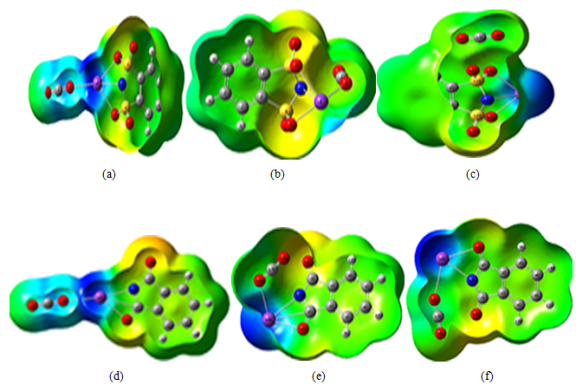
Figure 9. The ESP map of C6H4KNS2O4-CO2 and C8H4KNO2-CO2 complexes calculated at M062X/6-31+G(d,p) level in gas phase.
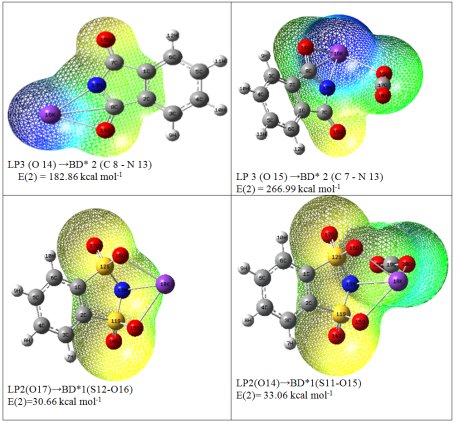
Figure 10. The schematic graphs of the charge transfer occurring from the lone-pairs to the anti-bonding orbital based on the NBO analysis at M062X/6-31+G(d,p) level.
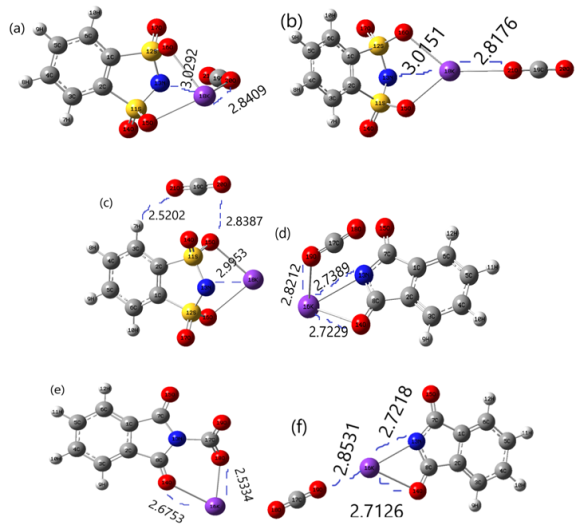
Figure 11. Solvents effect on the geometries of (a-c) and (d-f) complexes of C6H4KNS2O4 and C8H4KNO2 with CO2 were calculated by polarized continuum model (PCM) at M062X/6-31+G(d,p) level.
Information

